





What will pandemic disruptions mean for EU IVDR?
As the date of application for In Vitro Diagnostics Regulation (IVDR) remains on track for May 2022, regulators, industry and notified bodies (NBs) have many considerations to prepare for over the coming months. The coronavirus pandemic could add to an already tight deadline.
Alongside the EU’s Medical Device Regulation (MDR), IVDR provides a harmonized regulatory framework to ensure the safety and performance of devices, putting greater responsibility on manufacturers to demonstrate that products have met the requirements to be placed on the market by performing conformity assessments. IVDR also brings increased oversight to the in vitro diagnostic industry, including an expanded scope of requirements for performance, design and manufacturing, encompassing greater risk-control measures and heightened metrological traceability. Manufacturers also face pressure to quickly develop rapid and at-home diagnostics to support the COVID-19 crisis.
According to MedTech Europe, 85% of the more than 50,000 IVDs currently on the market will require certification by a designated NB in less than 20 months, which presents a considerable challenge in meeting the current deadline. The European trade group has urged regulatory authorities to delay IVDR due to the COVID-19 pandemic to no avail.
IVD manufacturers that have not begun preparations for IVDR urgently need to secure a strategy to address gaps in meeting the new regulation to avoid potential EU product availability issues. Here, we discuss steps that can be implemented to ensure a successful transition by the May 2022 deadline.
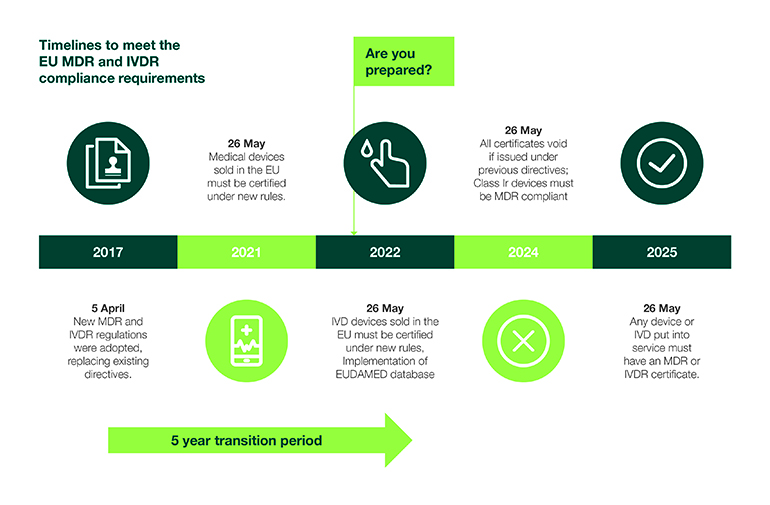 Assess product portfolios, quality systems over the IVD development lifecycle
Assess product portfolios, quality systems over the IVD development lifecycle
The IVDR affects the entire IVD lifecycle, and additional requirements impose significant increases in workloads. As manufacturers begin to calculate potential roadblocks and timeline constraints, market access priorities may begin to shift as a result. As there is no grandfathering of products, manufacturers must conduct an extensive gap assessment of their entire product portfolio for the purpose of evaluation and prioritization. Gap assessments should consider scope, sales, new product pipeline, financial impact and resources. Performing a gap analysis is integral to helping maintain and build market share through the transition.
While evaluating product portfolios, manufacturers will also need to:
• Assess current specifications and standards including requirements for regulatory purposes, such as quality management systems (QMS).
• Establish a transition plan for updated processes driven by IVDR.
• Bolster quality systems to allow for emergency use processes driven by diagnostic development associated with the pandemic.
Next, manufacturers will need to implement the transition plan through updates or creation of new standard operating procedures to comply with the IVDR. These operating procedures should incorporate several elements of medical device ISO standards, including the requirement that manufacturers operate a QMS. This would include device-specific design and development information, such as production and service controls, risk management and control measures, performance in alignment with intended use, and product interoperability and compatibility. They must also assess new roles for economic operators (e.g., manufacturers, authorized representatives, importers and distributors who share the duties of ensuring compliance) and persons responsible for regulatory compliance evaluation.
Plan and implement technical file remediation
After assessing portfolios and identifying gaps, manufacturers should define a remediation plan to create the required records for compliance. Once they implement a plan, manufacturers will need to update not only their QMS but also their design history file (DHF), which contains the records necessary to demonstrate that the design was developed according to approved design plans. In addition, they will need to develop a device master record, which comprises all the instructions, drawings and other records that must be used when producing a product.
If applicable, manufacturers will need to create periodic safety update reports (PSUR), post-market surveillance (PMS) plans and reports, post-market performance follow-up (PMPF), and a summary of safety and clinical performance. Lastly, sponsors will need to gather data to be submitted to the European Databank on Medical Devices (EUDAMED), as well as begin audit preparation.
Lastly, manufacturers will need to maintain their DHF, along with any technical files, while implementing PMS activities, including updates to PMPF and PSUR, in addition to data maintenance for the EUDAMED.
What lies ahead for 2021
For products in development and early in their lifecycle, manufacturers should ensure they are working towards IVDR requirement for EU approval. To maintain market share, manufacturers will need to address new clinical data, including technical file requirements; assess product portfolios for profitability, marketability and patient need; and finally, involve executive management and strategic partnerships in compliance efforts.
Angela Brown is global head and senior director of regulatory affairs, Icon. She has over 20 years of global regulatory affairs experience in medtech, specializing in international work with universities, start-ups and blue-chip companies.
Nicole A. Cowan is director of project management, IVD operations & strategy, Icon. She has over 15 years’ experience in clinical, academia, CRO, and central laboratory services.

