





Transitioning EU Clinical Trials: Key Steps for Compliance by 2025
EU Clinical Trials Transition
The conduct of clinical trials in the European Union (EU) and European Economic Area (EEA) has been regulated by the Clinical Trials Directive (CTD) since 2004. The CTD aimed for a coordinated approach to the regulation of clinical trials in all participating countries. However, this goal has been only partly achieved. Therefore, the European Commission (EC) determined to develop a new Regulation that would repeal the CTD, aiming to both simplify the submission of a clinical trial dossier for authorization and harmonize the requirements for conducting clinical trials.
The Clinical Trials Regulation (EU) No 536/2014 (CTR) was already adopted in 2014. However, one of the requirements of the CTR is the use of a newly developed, single electronic submission portal called Clinical Trial Information System (CTIS). Due to a delayed technical development of this portal, the CTR officially came into force on the 31st of January 2022, together with the go live of CTIS. From that date on, the CTR repealed the CTD.
Transition from Clinical Trials Directive (CTD) to Clinical Trials Regulation (CTR)
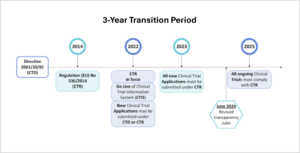
The main goals of the newly implemented CTR and accompanying CTIS portal are to further streamline processes, enhance transparency, and improve the overall efficiency of conducting clinical trials in the EU/EEA. To facilitate a smooth transition from the CTD to the CTR requirements, a three-year transition period (Figure 1) was established:
- Phase 1: January 31 2022 – January 31 2023
The CTR is in force and the accompanying CTIS portal is live. Sponsors could however choose to submit new clinical trial applications either still under the CTD, or already in compliance with the CTR. - Phase 2: January 31, 2023 – January 31, 2025
All new clinical trial applications must be submitted via CTIS and comply with the CTR. The CTD can no longer be used for new trials, but ongoing trials, approved under CTD remain to be governed by the CTD. - Phase 3: January 31, 2025 – onwards
This date marks the end of the transition period. All ongoing clinical trials that were approved under the CTD must now fully be transitioned to comply with the CTR. If not transitioned by this date, this will have serious consequences. Such trials are considered non-compliant and in breach of the CTR, meaning that sponsors could be subject to corrective measures and penalties by Member states, and subject to civil and criminal liability.
Source: Celegence
Clinical Trials in Scope of Transitioning
Not all clinical trials that have been approved under the CTD need to be transitioned to comply with the CTR. Transition is only required for CTD clinical trials that:
- have an expected completion date after 30 January 2025
- have at least one active site within the EU/EEA after 30 January 2025
- will be conducted in an additional Member State, for which no CTD application was submitted before 31 January 2023
Note that trials for which an end of trial notification has been submitted in all participating EU/EEA Member States, but the global end of the trial has not been notified, are not in scope to be transitioned. Furthermore, if a clinical trial has started prior to the CTD (in 2004), such trial should not be transitioned. Such trials should be submitted as a new clinical trial application under the CTR in CTIS.
Main differences between CTD and CTR
The first important difference is in the name: a directive versus a regulation. Both directives and regulations are issued by the EC; a directive needs to be transposed into national legislation, while a regulation is binding in its entirety and immediately applicable in all Member States without national modifications. The replacement of the directive by a regulation is therefore an important step in facilitating harmonization across all Member States. Other key differences between the CTD and CTR include the application and assessment processes (including substantial modifications), transparency rules and safety reporting.
Application and Assessment Processes in EU Clinical Trials
Under the CTD, a separate clinical trial application had to be submitted to each participating Member States. As harmonization and simplification of the regulatory processes regarding clinical trials are important pillars of the CTR, this principle has changed. The CTR follows a centrally coordinated approach, where the clinical trial application is submitted only once via the CTIS portal. A clinical trial application in CTIS consists of two main parts: Part I and Part II. Part I contains study specific documents, like trial details, sponsors and product sections. Part II contains all country/site specific documents, including, for example, trial sites and documents related to privacy, insurance, and patient information. For each clinical trial submitted in CTIS, one Reporting Member State leads the assessment process for those elements that are common throughout the EU. This means that there is a single assessment for Part I, but each concerned Member State is assessing Part II individually. As a result, discrepancies and delays can be reduced, and the approval process becomes more streamlined.
Substantial Modifications Under CTR
Like for initial clinical trial applications, also substantial modifications needed to be approved by each Member State separately under the CTD. With the CTR, all modifications need to be submitted via CTIS. The assessment of Part I modifications is coordinated by the reporting Member State, and for Part II modifications the concerned Member State(s) will perform the assessment.
Transparency Enhancements in Clinical Trials
The public access to trial information and results was very limited under the CTD. One of the goals of the CTR is to increase transparency, which is achieved by publication of key documents on a publicly available workspace on the CTIS website. While protecting personal data and commercially confidential information, publicly available information will contribute to the protection of public health and foster the innovation capacity of European medical research.
Recently, on 18 June 2024, revised CTIS transparency rules were implemented to lower the administrative burden for clinical trial sponsors, while making it easier for the general public to find relevant information on clinical trials. This revision was the result of feedback provided by users of the CTIS portal (including sponsors, clinical research organizations, academia, national competent authorities, ethics committees, health care professionals, and patients organizations), who requested an improvement of users’ experience and easy access to the relevant clinical trial information.
An important revision regarding transparency, is the removal of the deferral mechanism to delay publication of clinical trial documentation. On the other hand, however, there is a reduction in the amount of documents in scope of publication to only those documents that are essential to patients, healthcare professionals and researchers. An overview of documents and structured data fields in scope of publication, including the corresponding timing of publication, can be found in Annex I of the Revised CTIS Transparency Rules. Documents in scope of publication require two versions to be uploaded in CTIS: a version ‘for publication’ in which all personal data and commercially confidential information is redacted, and a non-redacted version ‘not for publication’, which is only used by the health authorities for their assessments. Altogether, these changes will result in earlier publication of key documents and a simplification of the CTIS portal.
Safety Reporting Requirements
Under the CTD, the safety reporting requirements regarding clinical trials differed across Member States. With the introduction of CTIS under the CTR, there is a central place for annual safety reports, which will be assessed by a so called Safety assessing Member State (saMS). The saMS will carry out the assessments for all investigational medicinal products containing the same active substance, regardless of the pharmaceutical form, strength or indication, and regardless if they are used in several trials with different sponsors. This means that a Member State can be a saMS, even when the trial is not conducted in that Member State. After the assessment, the saMS shares its findings with all Member States.
Key Actions for Transitioning Clinical Trials to CTR
Since the end of the 3-year transition period is approaching, it is of utmost importance that sponsors of clinical trials that were approved under the CTD and not yet transitioned to comply with the CTR, take action. The main steps that sponsors should take are listed below.
Assess Current Clinical Trials for Transition
With the limited time remaining for transitioning clinical trials, sponsors should already have performed a thorough review of all ongoing clinical trials to determine their status and the necessity to apply for a transition to the CTR.
Familiarize with the Clinical Trial Information System (CTIS)
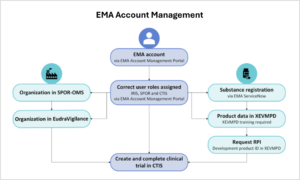
Online training modules and guidance are available to train relevant staff members on the new portal for the submission and management of clinical trial data. EMA also organizes live events to support clinical trial sponsors in becoming familiar with CTIS and the processes required to transition ongoing clinical trials (e.g. webinars, Walk in Clinics, Bitesize talks). Keep in mind that there are several interdependencies with different EMA systems required for the use of CTIS (Figure 2). These include having an EMA account with the required roles (EMA Account Management), the registration of your organization in EMA’s Organization Management Service (OMS), and registration of your investigational medicinal product into EMA’s Extended EudraVigilance medicinal product dictionary (XEVMPD).
source: Celegence
Update Clinical Trial Documentation for Compliance
Update all clinical trial documentation to comply with the CTR requirements. As trials in scope of transitioning already received approval under the CTD, there is no in depth assessment required. To ensure a fast procedure for transitioning such trials, a minimum set of information is required for the initial transition application:
- All mandatory application data fields in CTIS
- Statement on compliance with the Global Data Protection Regulation (template)
- Cover letter (template)
- Latest authorized version (under CTD) of:
- Protocol (harmonized or consolidated)
- Investigator’s Brochure (IB, harmonized or consolidated)
- Investigational Medicinal Product Dossier (IMPD, harmonized or consolidated)
- Good Manufacturing Practice relevant documents
- Documents related to auxiliary medicinal products (called non-IMPs under CTD)
- Subject information sheet and informed consent form
As CTIS has a fixed structure with all kinds of mandatory fields and documents to be uploaded, a sponsor may submit placeholder documents for the documents not listed as part of the minimum dossier. Provide the following text in such placeholder documents: ‘This aspect was assessed by National Competent Authority (NCA) and/or ethics committee who has given a positive opinion on the clinical trial under the CTD and is therefore covered by the conclusion of the assessment under the CTD’. These documents shall be updated at time of the first substantial modification, to complete the clinical trial application in compliance with the CTR rules. Note that only documents which are affected by the substantial modification can be updated.
Since separate clinical trial applications had to be submitted to each Member State under the CTD, there may be differences is key documents like the protocol, IB or IMPD of multinational clinical trials. Before transitioning such trials, it is required to have a harmonized or consolidated version of these documents, for which the Clinical Trial Coordination Group (CTCG) has published a best practice guide.
Prepare and Submit Regulatory Submissions
Prepare and submit the necessary documentation to transition trials to the CTR. There is an agreement amongst Member States that transitional clinical trial applications submitted before 16 October 2024 will fall under an expedited transition procedure, focusing on validation (instead of assessment) of the minimum dossier of CTD approved documents. If submitted before this deadline, completion of the transition before the end of the transition period can be assured.
Be aware that it is not possible to submit a transition application when there are ongoing substantial modifications in any of the concerned Member States under the CTD. Considering the short time left for the transition of ongoing clinical trials, it is recommended to withdraw any CTD substantial modifications that do not have a decision yet, and instead focus on submitting consolidated Part I documents directly.
Important Deadlines and Considerations for EU Clinical Trials
- End of transition period is marked by the 30th of January 2025. After this date, all ongoing clinical trials must comply with the CTR.
- Approval of a transition application may take up to 3 months. Take into account the 2week winter clock-stop at EMA.
- Member States agreed on a deadline for submitting transition applications for an accelerated assessment. This deadline is 16 October 2024.
- Planning is key! It is not possible to transition a clinical trial when there are other ongoing assessments, like for substantial modifications.
- EMA provides a lot of information and support for sponsors of clinical trials. It is recommended to subscribe for the CTIS Newsflash, which provides key updates on the latest developments, and links to useful reference materials.
Article source: MedTech Intelligence

